CerebelloGenesis
Launched in the Forthcoming Special Issue of "The Cerebellum"

Guest Editor: Dr. András J. Pellionisz:
Cerebellar FractoGene - Genesis and Pathogenesis of a Neural Network for Spacetime Coordination
An Editor of "The Cerebellum", Dr. András J. Pellionisz will be the Guest Editor of a Special Issue, putting together a span of genome-to-overall sensorimotor space-time coordination by the Cerebellum. A Special Issue of the peer-reviewed Internet Journal "The Cerebellum" will be issued (with the Chief Editor of the Special Issue), per request of the Chief Editor of The Cerebellum Journal.
The Cerebellum is the first and perhaps most unique part of the CNS whose function is explained in terms of advanced mathematics

as well as the emergence of the Neural Network has started to be explained in terms of fractal geometry (look at the Purkinje neuron emerging from a fractal set).
This initiative will re-vitalize all Neural Net efforts, by bringing them - via the Cerebellar Neural Network - into Genomics.
FractoGene introduces a novel concept of evolution that is the opposite to non-coding sequences interpreted as "evolutionary leftovers". The primary "inventions" of Mother Nature are the genes (coding DNA sequences). Later, evolution is "double threaded". On one hand, there is a steady but slow addition of genes (each determining a fractal template), while the secondary "improvements" are the instructions encoded in the repetitious introns, determining e.g. how many "generations" of fractal growth should occur to generate an organ(ism) that is higher in the evolutionary ladder. This is totally consistent with the facts that primitive organisms (bacteria) have no introns at all, while the higher we go on the evolutionary ladder, the higher percentage of the total DNA is composed of "junk DNA" (appropriately called fractal repetitions). The cerebellar Purkinje cell is an excellent example. The "gene" of Purkinje neuron bifurcation is present throghout the evolution (starting with the sharks and up), while it is a fact of anatomy that Purkinje cells are very simple in lower ranking animals (frogs, needing just 2-3 generations of bifurcations, defined by 2-3 introns how the gene should manifest itself differently at the higher branch(let)s). The Purkinje neuron of the guinea pig (pictured in the example) has a higher number of bifurcation-generations, while the cat cerebellum is known to have Purkinje cells that is more complex. The human Purkinje neuron is even more advanced, needing more fractal generations of bifurcations - hence there is the highest percentage of introns. This explanation is also totally consistent with the rather astonishing fact (known for the last few weeks only), that the gene pool of the mouse is 99% identical to that of humans, while the total DNA of the mouse is only about 2.5 billion base-pairs, while the human DNA is over 3 billion. What makes us so vastly different from the mouse, is the fractal generations, determined by the vastly higher percentage of "non-coding DNA" (loosely speaking, "introns").
The Cerebellum puts the "junk DNA" theory to rest also in a very different, "global structuro-functional" manner.
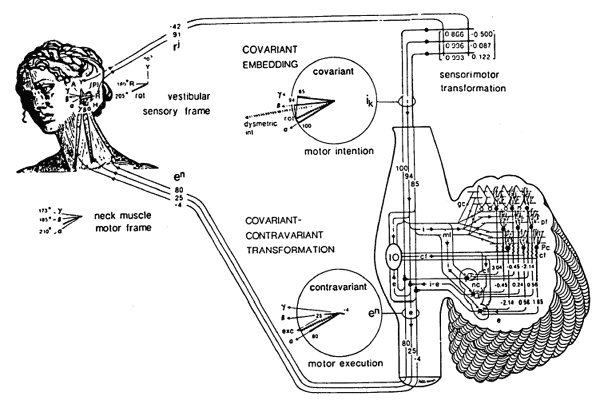
Also, the Cerebellum is thoroughly investigated over several decades, and has been modelled mathematically at overall functional, neural net morphogenesis, neural net function, cellular, cell membrane levels - and now also at the level of genomics. The cerebellum is perhaps the best known part of the CNS, whose function (sensorimotor spacetime coordination) is known both in terms of physiology, morphology and pathology, as well as there are mathematical explanations (PL 1980, 1982, 1985,,summed up in Pellionisz 1987, award-winning in 1989), producing quantitatively testable predictions that have been tested and verified) how the neural network of only 5 types of neurons produce this brain function (explains coordination by means of generalized coordinates, tensors).
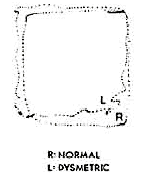
Lack of cerebellar function is known as "ataxia" (or dysmetria - lack of metric). For instance, Fig. 5. in Pellionisz & Llinas 1980 predicts the "ataxic" motor execution of handwriting in case of cerebellar lesion, to be compared with physiological observations (Fig. 2. in Pellionisz & Llinas 1980).
It represents a major leap that cerebellar ataxia has lately been directly linked with an identified "run of an intron", a tremendous discovery by Massimo Pandolfo (see below). Therefore, the Cerebellum is emerging as an organ whose function can now be described by Information Geometry (Tensor Nework Theory), their primary neuron (the Purkinje cell) can be modeled by Fractal Geometry, and its genetic determination by FractoGene (Fractal DNA Geometry).
1996
Massimo Pandolfo
Identifies the 1st intron of a gene on chromosome 9 (both in humans and in mouse model). Normally, the intron of the gene is repeated 7 times. In same cases (1:50,000) there is a "run" on this intron, sometimes into the thousands of repetitions. This causes spinocerebellar ataxia and cardiomyopathy. Clearly, the repeated introns are not some useless, evolutionary leftover "junk" - but play a crucial role (in this case on the iron household regulation in mitochondria). With this discovery, the "theory" of "junk DNA" is directly contradicted by experimental (clinical) evidence. However, "facts don't kill theories" - only "better theories kill less superior theories". FractoGene, that explains the role of intron-repetitions in terms of fractal geometry; i.e. how many times a fractal self-similar repetition must occur for healthy growth, killed the "Junk DNA Theory".
Details from the webpage announcing Massimo Pandolfo's discovery
Scientists have identified a new type of trinucleotide repeat mutation that leads to Friedreich's ataxia (FA), a rare childhood neurodegenerative disease. The discovery allows accurate screening for carriers of the disease and may lead to the first effective treatments.
The mutation contains between 200 and 900 repeats of a normal three-base (trinucleotide) sequence -- guanine, adenine, and adenine, or GAA -- in a newly described gene called X25. The normal gene contains only seven to 22 repeats. Mutated X25 genes produce much less messenger RNA, an intermediate step in protein production, than normal genes.
X25 normally codes for a protein called frataxin that is concentrated in the organs affected by FA, says Dr. Massimo Pandolfo of Baylor College of Medicine in Houston. Frataxin is unlike any previously described protein, and scientists are just beginning to learn how it works. Pandolfo conducted the study in collaboration with scientists from France, Spain, and Italy. The work was funded by the National Institute of Neurological Disorders and Stroke (NINDS) and by the Muscular Dystrophy Association. It appears in the March 8, 1996, issue of Science.*
About 98 percent of FA carriers have the expanded GAA sequence, so scientists can now diagnose carriers very accurately, Pandolfo says. Studying how X25 and frataxin function also will help researchers understand the disease and design treatments for it, he says.
FA affects several thousand people in the United States and causes degeneration of the spinal cord and its brain connections, the heart, and the pancreas. Symptoms typically appear between the ages of five and 25 and include progressive loss of coordination (ataxia), lack of leg tendon reflexes, and speech slurring. Most patients also develop heart enlargement, and about 10 percent acquire diabetes mellitus. At present there is no treatment to slow progression of the disease, and patients eventually must rely on a wheelchair. They also have a shortened life expectancy, averaging 37 years.
Unlike mutations in other trinucleotide expansion diseases, including Huntington's disease, the FA mutation is genetically recessive. This means a child must inherit two mutated copies of the gene -- one from each parent -- to develop the disorder. Preliminary results suggest that, in contrast to other trinucleotide expansions, the number of repeats in the FA mutation does not correlate with disease severity or age of onset. People carrying only one altered copy of the gene do not have FA but can pass the mutation to their children. Previously described trinucleotide repeat mutations are dominant, meaning only one altered copy of the gene leads to disease.
Since carriers of FA are themselves unaffected by the mutation, they are usually unaware of it and tend to have the same number of children as other people. This accounts for the relatively high frequency of carriers in the population -- about one in 100 people, according to the National Ataxia Foundation.
The FA trinucleotide expansion is the first disease-causing mutation found to occur in an intron. Introns code for sequences that are cut out of messenger RNA before proteins are assembled. Scientists have long wondered what function introns serve. The FA mutation supports previous evidence that they influence gene expression.
Along with the trinucleotide expansion, Pandolfo and his colleagues identified three single-nucleotide "point" mutations that inactivate the X25 gene and lead to disease. These mutations affected only eight of the 184 FA patients examined. These patients all had the expanded GAA repeat on the X25 gene from their other parent. "Patients with FA either have both genes with the unstable expansion, or they have one gene with the unstable expansion and the other with a point mutation," says Dr. Giovanna Spinella, a pediatric neurologist at NINDS.
While frataxin's function is still unclear, it includes a 27 amino acid sequence that is nearly identical to sequences from a worm species (C. elegans) and yeast (S. cerevisiae), Pandolfo says. The similarity of this sequence in such diverse species, which are separated by millions of years of evolution, suggests that it plays a critical role in organisms' survival. The rare "point" mutations all affect this region of the protein, underscoring its importance.
Studying how frataxin works in other species may help scientists learn what it does in humans and design treatments for FA, Pandolfo says. He and his colleagues are now working to further define frataxin's function. This work may reveal new protein interactions that are important to all cells, not just those affected by FA.
FA is the eighth disease linked to trinucleotide repeat mutations. The others are Huntington's disease, spinocerebellar ataxia type 1 (SCA1), X-linked spinobulbar muscular atrophy (Kennedy's disease), dentatorubral and pallidoluysian atrophy (DRPLA), spinocerebellar ataxia type 3 (SCA3 or Machado-Joseph disease), fragile X syndrome, and myotonic dystrophy. FA is the first disease linked to an expanded GAA sequence.
Already published article in this e-Journal is the following, sheding light on the Genomic base of Cerebellar Function:


The above 8kb of information is fully mapped both in human and in the mouse, yet "genome data bases" strip the "junk DNA" and give you only the exons (in this case, the combined 465 base-pairs):
BASE COUNT 107 a 156 c 119 g 83 t
ORIGIN
1 taggtactag gatttagggg cacttctgag ccccatttcc ctggcaggtt caccggacca
61 ggaaggcttc ttcaacctgc tgacccacgt gcagggcgat cggatggagg agcagcgctg
121 ttccttgcag gctgggccag gccagaaccc agaaagccag ggtggccctg ctccagagat
181 ggacaatctc atggatatgc tggtcaacac ccagggccgc cgcatggacg accagcgtgt
241 aacagttaat tccctgcctg gcttccaacc tatcggcccc aaggatggaa tgcagaaacg
301 acctgggacc ctcagccctc aacccctgct cacccctcag gatcctgctg cactcagctt
361 ccgcaggaac agcagccccc agccccagac acaagctcct tgagagttct agccatcctg
421 ggcctcccac tggcccctga aaacaataaa acacttggca ctagc
PROTEIN TRANSLATION:
"MEEQRCSLQAGPGQNPESQGGPAPEMDNLMDMLVNTQGRRMDDQ
RVTVNSLPGFQPIGPKDGMQKRPGTLSPQPLLTPQDPAALSFRRNSSPQPQTQAP"
INFORMATION TECHNOLOGY CHALLENGE:
With 8kb of PCP2 only 465 basepairs coding - most databases don't even contain (much less analyze!) the "non-coding part" of the DNA. Analysis of this DNA information, especially of its repetitive segments, this small but distinguished part of the genome, could be a key for a paradigm-shift. By now, it is known that some sequences of the DNA are protein-coding, others are not. Metaphorically, it is similar to 100101... (etc) binary sequences in traditional computers, where some contents of the memory are "instructions", other sequences are "data" - and it is impossible to tell which is which (see the original concept of computers by Johnny von Neumann).

(from literature)
For the reader's benefit, we emphasize that his is only a metaphorical reminder to expect "unreasonable" or "nonsensical" segments of a code. In the case of DNA, we are dealing with a pack of biological information, that we propose is encoded not as in computers but as in fractals.
Nonetheless, part of the information, the "genes" (protein coding sequences), although essential, may only tell the basics of the story. With higher ranking species, the repetitious "non-coding" sequencess may "just" spell out how the fractal template (determined by the coding sequences) should be different in the higher and higher generations of the expression.
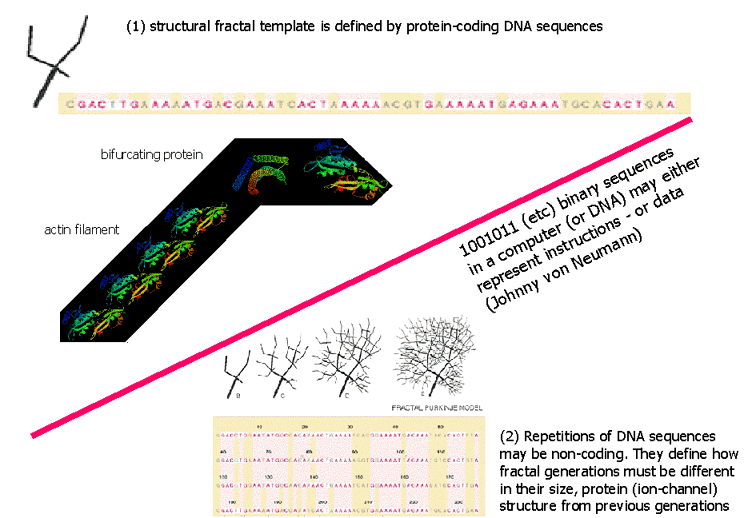
The basics of the "protein coding" DNA sequences are undisputable. For instance, in case of the Purkinje cell (and a great number of cells) the protein named "actin" forms "actin filaments", to determine the cytoskeletal structure of the cell:
 Fractal Organelles: Mitochondria . Do proteins show fractal properties?
Fractal Organelles: Mitochondria . Do proteins show fractal properties?
Cited Schematic Figure of Cellular Structure from Literature:
{kind=link}

Fractal Model of the Mitochondrium by Jules Ruis
Cytoskeletal proteins (actin filaments as well as cytoskeletal profilins and other "bifurcating" proteins) determine the basic shape of the neuron.
These "actin filaments" are well documented to determine the structure of the cell. For "actin filaments" see e.g. the diagram of Fig. 4. in "The p75NGF receptor mediates neuronal survival in vitro and in vivo" , and in vivo picture. For a diagrammatic "actin filament" see schematic Figure.
CEREBELLAR (PATHO)GENESIS
As indicated above, the cerebellum is not only a beautiful example of our knowledge reaching over boundaries of traditional morphology and physiology (and at this point, also genomics), but it is also known that some hereditary diseases affect this stucture and function in a manner that is reasonably well understood already.
Friedreich' Spinocerebellar Ataxia (FA) is the eighth disease linked to trinucleotide repeat mutations. The others are Huntington's disease, spinocerebellar ataxia type 1 (SCA1), X-linked spinobulbar muscular atrophy (Kennedy's disease), dentatorubral and pallidoluysian atrophy (DRPLA), spinocerebellar ataxia type 3 (SCA3 or Machado-Joseph disease), fragile X syndrome, and myotonic dystrophy. FA is the first disease linked to an expanded GAA sequence.
The "program" outlined above lays down some basic elements of the "CerebelloGenesis", which is expected to cut across several species (certainly including humans, cats, rat, mouse, guinea pig) and might become the first comprehensive program to integrate "leading edge" results of genomics with more classical approaches.
SIGNIFICANCE OF UNLOCKING THE MOLECULAR MECHANISM OF CEREBELLAR (PATHO)GENESIS
Friedreich Disease (Spinocerebellar Ataxia) is a leading example of a very significant group of so-called trinucleotide diseases (Friedreich's, Alzheimer's, Parkinson's, Huntington's, Recklinghausen's being the most common diseases in this group). These neurodegenerative diseases (with complications as serious e.g. in case of Friedreich as lethal cardiomyopathy) attract much interest in funding. With an increasingly older populations, finding the cause at a molecular (DNA and protein) levels (see e.g. the "chimeric" frataxin) the practical outcome is potentially very significant. With both human and mouse DNA models available, the physiological as well as pathological cerebellogenesis could be pinned down, leading to a better understanding of Friedreich' Spinocerebellar Ataxia, and the rest of the trinucleotide diseases. Since the cause of this disease is a "run" on the introns, the CerebelloGenesis project differs in two major aspects from the trends prevailing thus far
1) Instead of aiming at a "full genome" sequencing of a single animal, it directs attention, across species, on a single, well-distinguished organ (the cerebellum).
2) Instead of solely targeting "genes" (protein coding DNA sequences) it directs attention to the "non-coding sequences" (98.7% of the DNA).
In addition, the Neural Net program of the Cerebellum already showed once that an understanding of the mathematical (information technology) basics of the Cerebellar Neural Net could lead to useful applications as large-scale as Flight Control of F15 Figther Planes for NASA. A program was elaborated for NASA-NIH-NIMH-NSF in the course of 3 months and NASA flew an F15 with "artificial cerebellum" within a few years.
Information Technology applications, once proven with Information Geometry of the Cerebellum, will help in the scale of pharmaceutical industry, to work out the Genome Geometry (based on FractoGene), leading to clues not only of a well-functioning Cerebellum, but also to a slew of dreaded diseases.
APPENDIX: TIMELINE
NEURAL MORPHOLOGY AND PHYSIOLOGY
First microscopical picturing of Cerebellar Golgi and Purkinje cells (Ramon y Cajal, 1911)
Nobel Prize for Camillo Golgi and Ramon y Cajal, 1906
First Nobel Prize for the Vestibulo-Cerebellar Apparatus (Robert Barany, 1914)
Nobel Prize for the DNA (Crick, Watson and Wilkins, 1962)
First comprehensive monograph on the Cerebellum (Eccles, Ito, Szentagothai, 1967)
Nobel Prize for Hodgkin, Huxley and Eccles, 1963
First computer model of a neural net (Cerebellum, Pellionisz 1970)
First anatomical large-scale modeling (Cerebellum, Pellionisz and Szentágothai, 1972)
First Hodgkin-Huxley compartmental Purkinje cell computer model (Pellionisz and Llinas 1977)
NEURAL GEOMETRY
First mathematical model of the cerebellar neural network function (Pellionisz and Llinas 1980, 1982, 1985)
First experimental verification of a mathematical cerebellar model with quantitative predictions (Gielen and Zuylen, 1986)
First application of the cerebellar modeling for the vestibulo-oculomotor reflex arc (1985)
First application of artificial cerebellum for aircraft control (1990)
First fractal model of a Purkinje cell (1989)
GENOME GEOMETRY
First mathematical (fractal) predictions for Purkinje cell DNA and its development in evolution (2002)